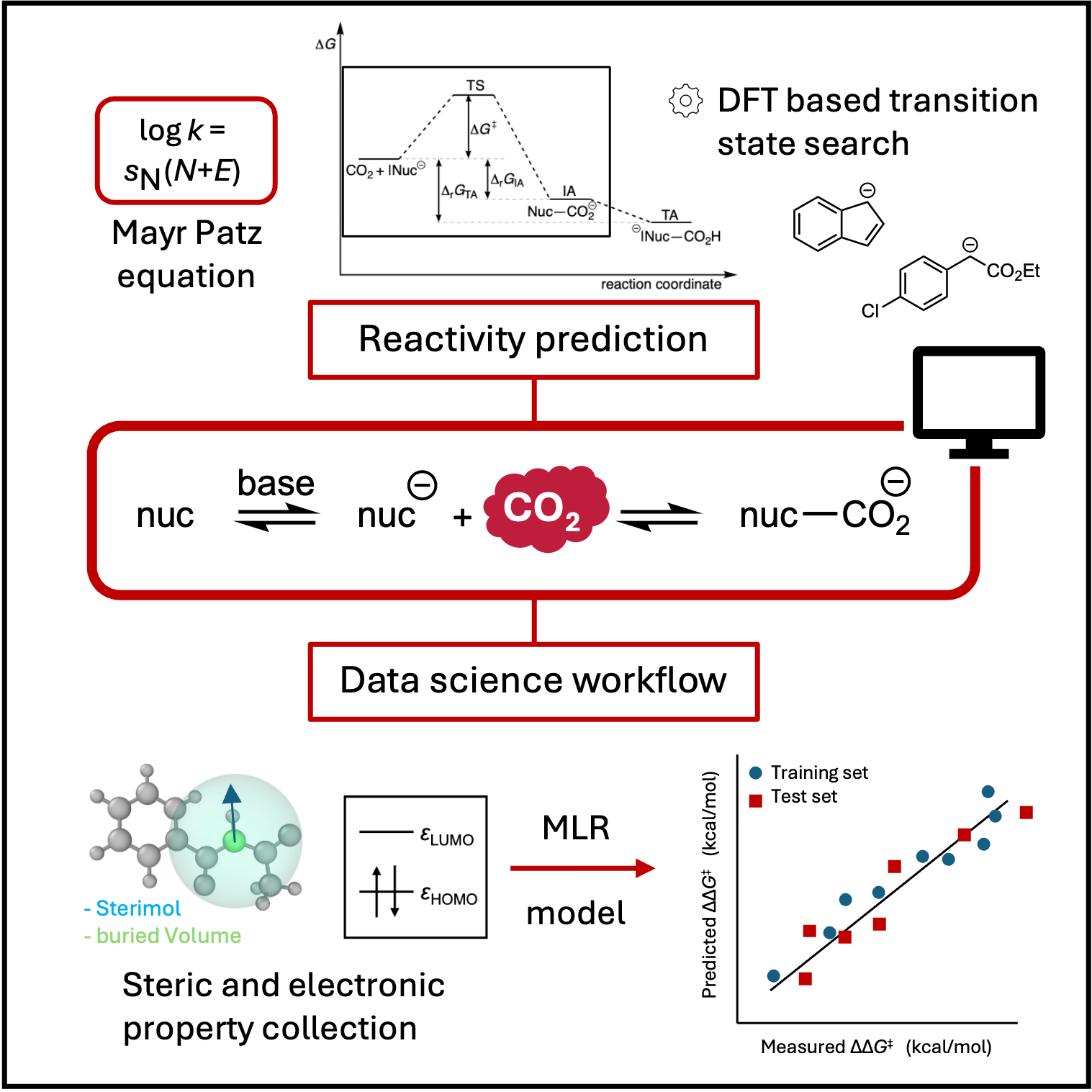
Chemische Reaktivität steht im Zentrum der Chemie. Durch die Verbindung von Quantenchemie und datengestützten Ansätzen wollen wir verstehen, wie und warum Moleküle chemische Umwandlungen durchlaufen – von den grundlegenden Prinzipien der Bindungsbildung bis hin zu weiterreichenden Fragen der Synthese und des nachhaltigen Moleküldesigns. Ein besonderes Augenmerk liegt auf gesellschaftlich relevanten Systemen wie Kohlenstoffdioxid, einschließlich der Stabilität seiner Produkte. Im datengetriebenen Teil unserer Forschung entwickeln wir Modelle zur Echtzeitvorhersage chemischer Reaktivität und chemischen Interpretierbarkeit, um eine routinemäßige Syntheseplanung zu ermöglichen. Ergänzend quantifizieren wir die Unsicherheit unserer Vorhersagen, um ihre Verlässlichkeit besser einschätzen zu können.
Leitende Forscherin: Maike Eckhoff
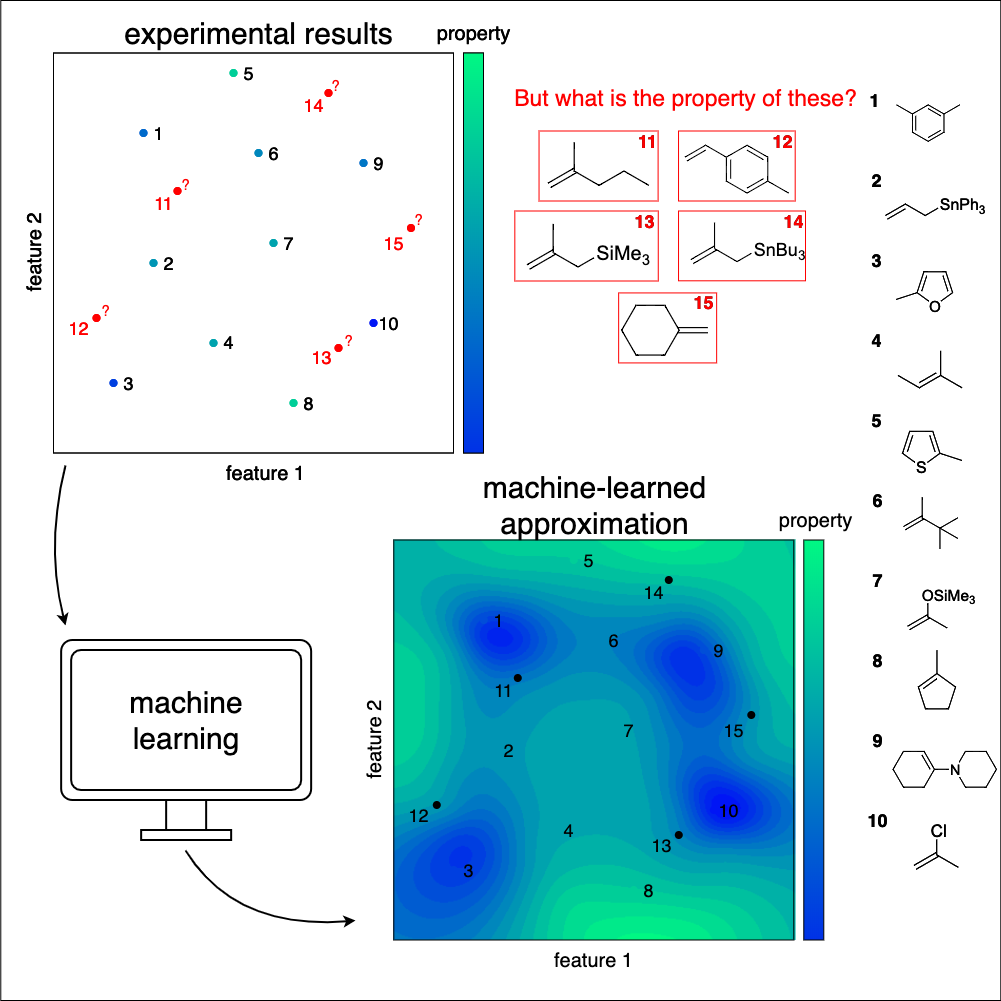
Active Learning steigert die Effizienz von Machine-Learning-Methoden, indem es selektiv die informativsten Trainingsdatenpunkte, die durch Experimente oder Simulationen erzeugt werden, bestimmt. Dadurch kann die Menge benötigter Trainingsdaten erheblich reduziert werden, was Ressourcen schont und die Effizienz ankurbelt. Dieser Ansatz ist insbesondere von Bedeutung, wenn Trainingsdaten knapp oder teuer zu beschaffen sind – wie in der Chemie oder den Naturwissenschaften im Allgemeinen.
Leitende Forscherin: Elizaveta (Liza) Surzhikova
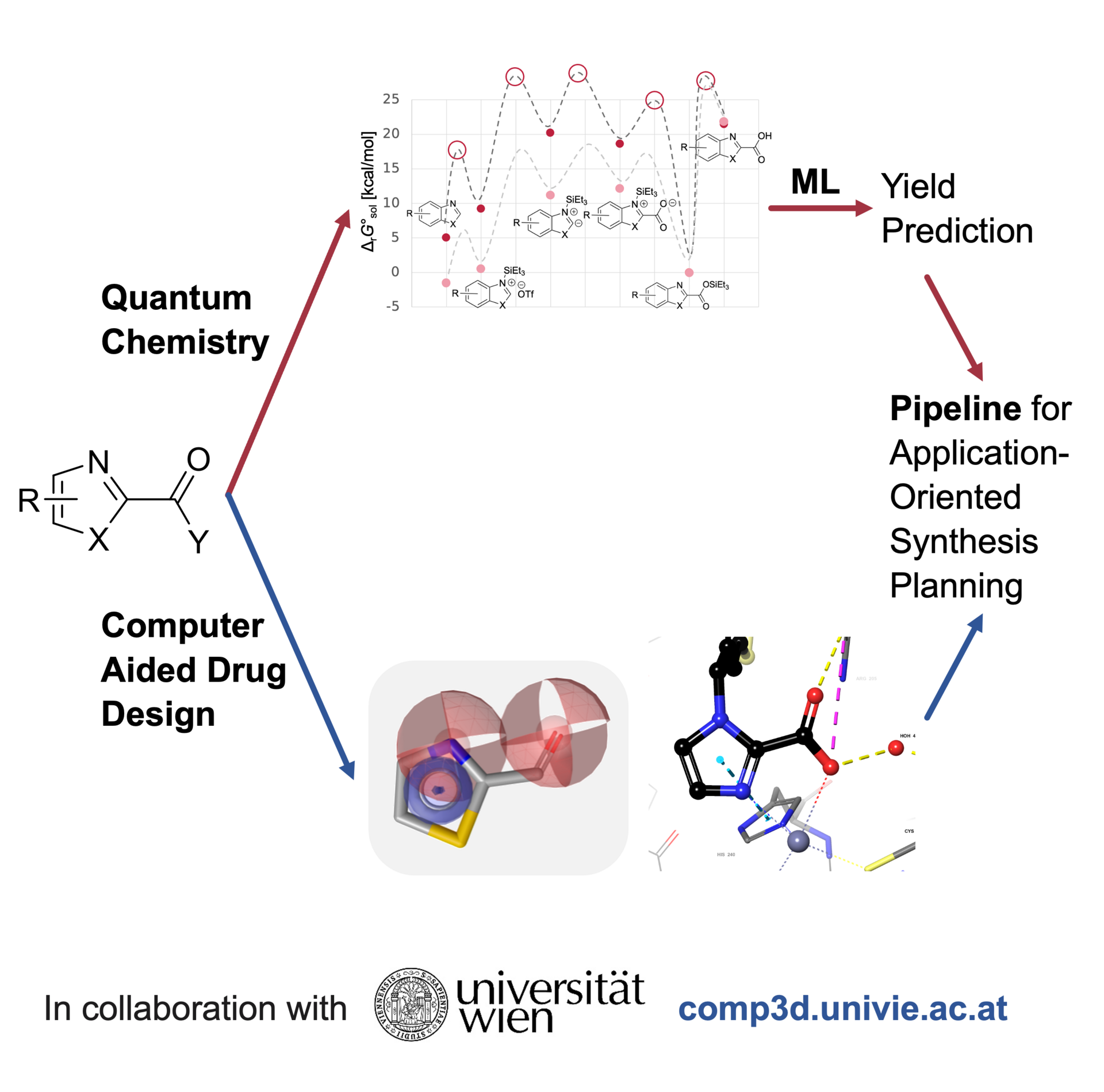
Wirkstoffdesign ist ein wichtiger Bereich der medizinischen Chemie, der sich auf die Entwicklung neuer therapeutischer Verbindungen konzentriert, die wirksam mit biologischen Zielstrukturen interagieren können, um Krankheiten zu behandeln. Die Synthetisierbarkeit dieser Moleküle stellt sicher, dass diese effizient und wirtschaftlich im Labor bzw. im industriellen Maßstab hergestellt werden können, was für die praktische Anwendung entscheidend ist. Durch die Integration von computergestütztem Wirkstoffdesign, Quantenchemie und Machine Learning wollen wir die Entdeckung neuer Wirkstoffe auf verschiedenen Ebenen beschleunigen.
Leitende Forscherin: Kerrin Janßen