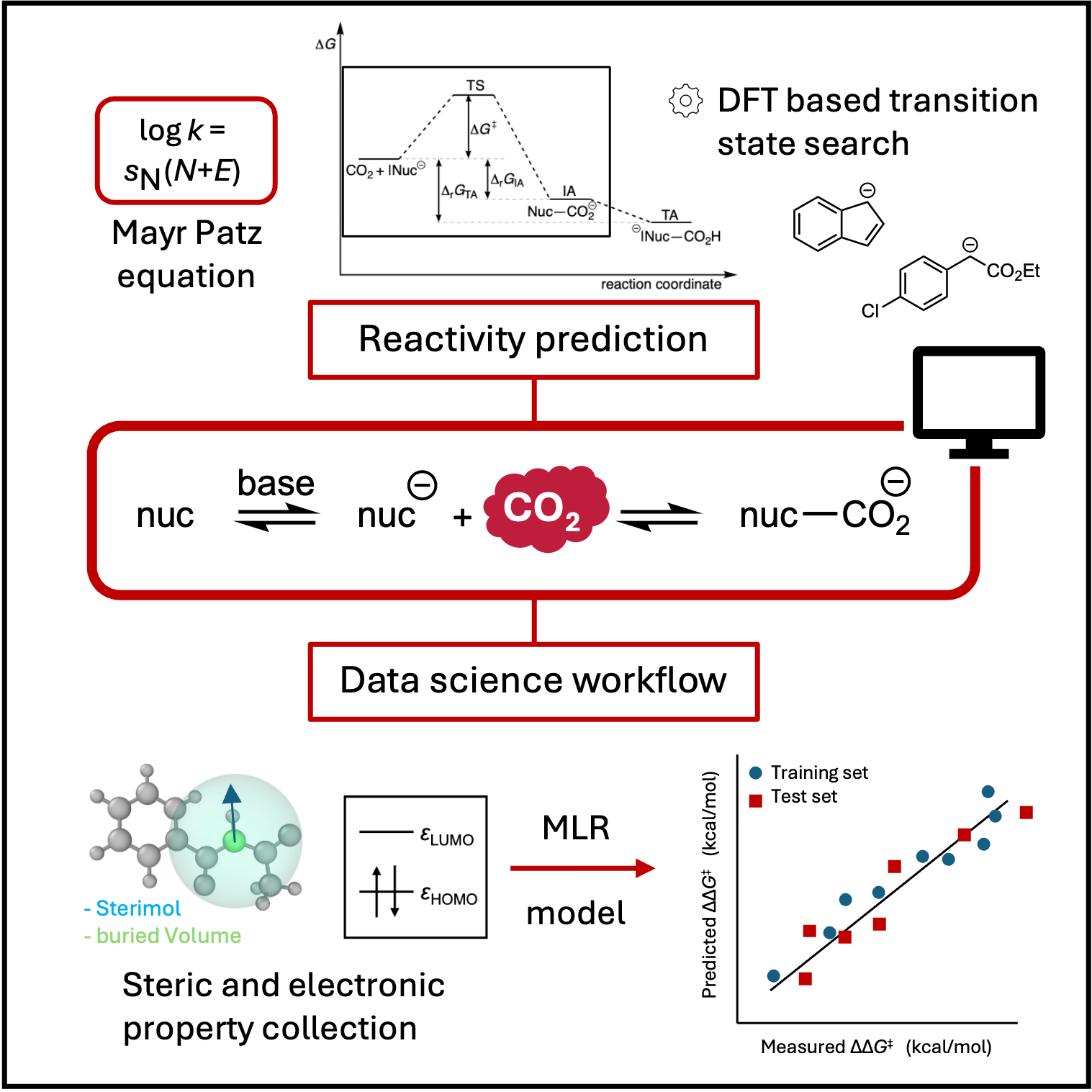
Chemical reactivity lies at the heart of chemistry. By integrating quantum chemistry and machine learning, we seek to understand how and why molecules undergo transformation—from the fundamental principles that govern bond formation to broader implications for synthesis and sustainable molecular design. Particular attention is given to systems of societal relevance such as carbon dioxide, including the stability of its products. On the data-driven side, we develop models for real-time reactivity prediction and chemical interpretability to enable routine synthesis planning, complemented by uncertainty quantification to assess model reliability.
Lead Researcher: Kerstin Bublitz
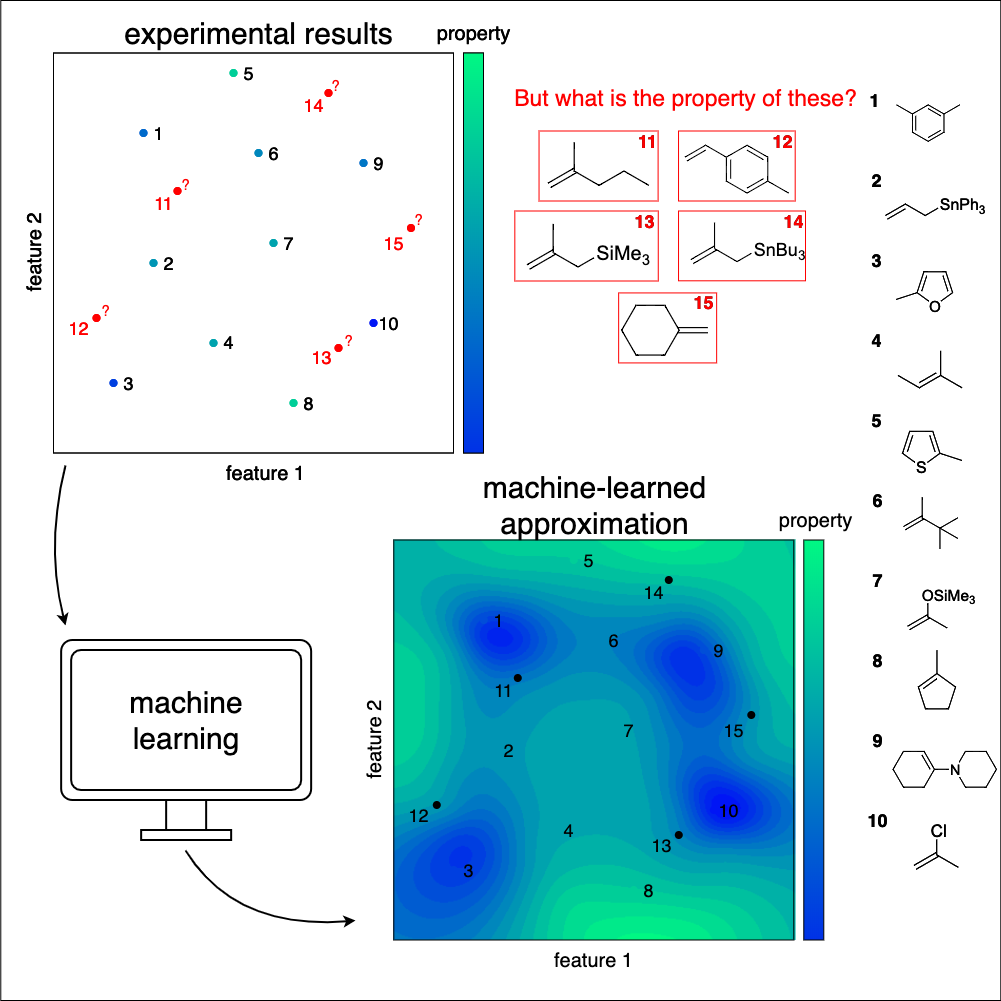
Active learning enhances the efficiency of machine learning workflows by selectively querying the most informative training data points generated by experiment or computation. In this way, the amount of training data needed can be significantly reduced, which saves resources and boosts efficiency. This approach is particularly important when training data is scarce or expensive to obtain—such as in chemistry or the natural sciences in general.
Lead researcher: Elizaveta (Liza) Surzhikova
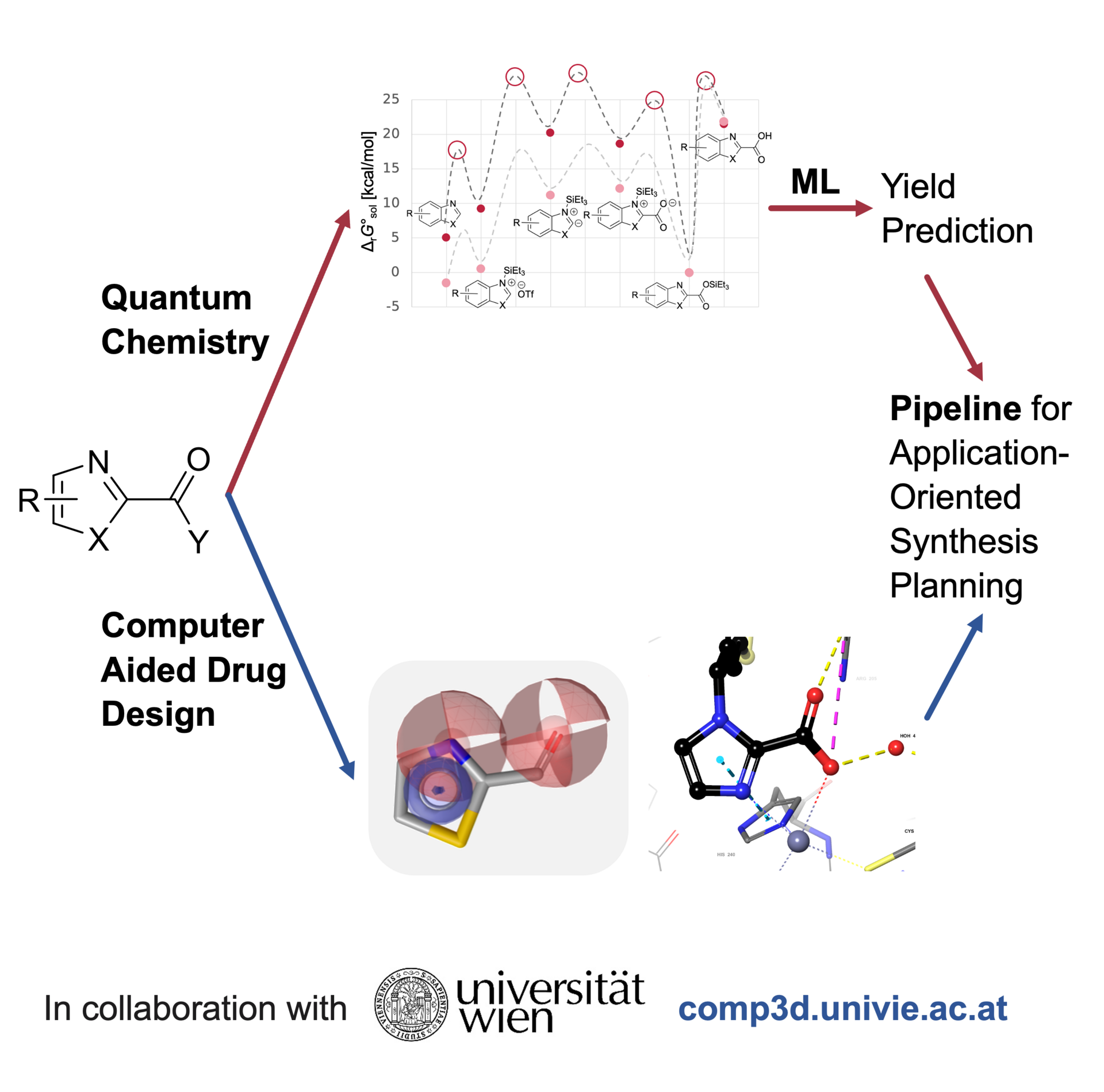
Drug design is a critical field in medicinal chemistry that focuses on creating new therapeutic compounds that can effectively interact with biological targets to treat diseases. Synthesizability ensures that the designed molecules can be efficiently and economically produced in the lab or on an industrial scale, which is crucial for practical application. By integrating computer-aided drug design, quantum chemistry, and machine learning, we aim to accelerate the discovery of new drugs on different scales.
Lead Researcher: Kerrin Janssen