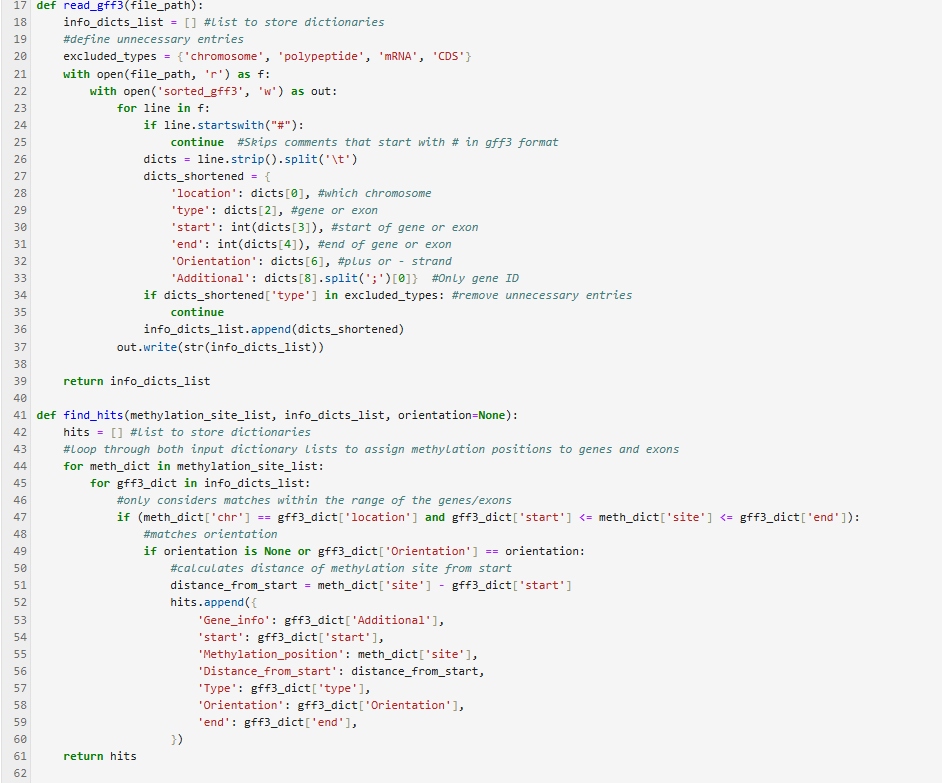
The Methylation Position Annotator identifies methylation on exons, introns, and promoters of genes and can be a useful tool in gene expression studies, as methylation of DNA can affect transcription. It works by comparing genomic methylation site positions from an input file to the locations of gene annotations saved in GFF3 file format, making it possible to identify methylated exons and introns. Additionally, it calculates the distance of the methylated position from the start of an annotated gene, which allows for the identification of promoters. The tool’s short run time and included input file filtering steps make it easy to get an idea of methylation in a genome of interest.
The code for the Methylation Position Annotator was written by Ronja Friedhoff.

With this code, it is possible to replace amino acids in aligned protein sequences with their corresponding codons from nucleotide sequences. This is essential for constructing phylogenetic trees based on nucleotide data, which better captures genetic variation and evolutionary relationships. Project completed by Luiza Galli.
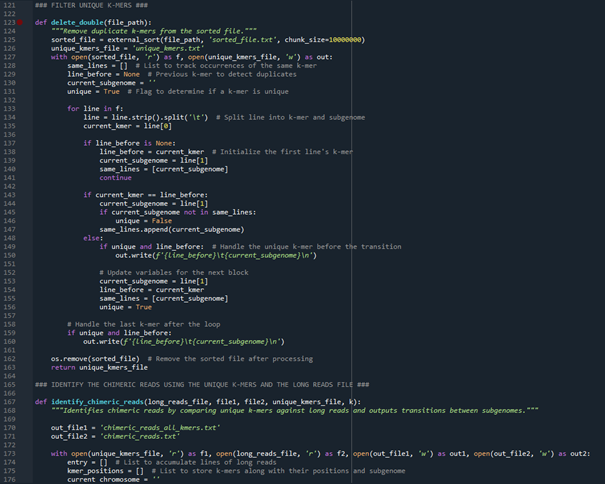
This code is able to identify homoeologous exchange events in polyploid plants based on long reads. For this purpose, two subgenome sequences of the plant are compared with each other to find unique k-mers for both subgenomes. These unique k-mers are then searched for in the long reads. If k-mers from both subgenomes occur in one read, it is stored as a chimeric read. A chimeric read indicates a homoeologous exchange event. The code works with FASTQ files for the long reads and with FASTA files for the subgenome sequences.
This project was completed by Mia-Sophie Brieske.