My research interests lie in the areas of preparative organometallic chemistry and coordination chemistry with an emphasis on the development of novel, unusual ligand systems and on the investigation and application of their transition metal complexes. During the last ca. 10 years, we have thoroughly studied the coordination chemistry of anionic imidazolin-2-iminato and neutral imidazolin-2-imine ligands [1]. In these compounds, the ability of the imidazoline moiety to effectively stabilize a positive charge results in a pronounced basicity of the exocyclic nitrogen donor atoms and in the formation of particularly strong metal-nitrogen bonds. These characterisitics qualify imidazolin-2-iminato ligands as ancillary ligands in homogeneous catalysis, and for instance, novel catalysts 1-3 for olefin polymerisation and metathesis were developed [2,3,4]. Other catalytic applications were realized with uranium complexes such as 4 (ring-opening polymerisation)[5] and with rare earth metal complexes such as 5 (hydroamination, hydrosilylation)[6].
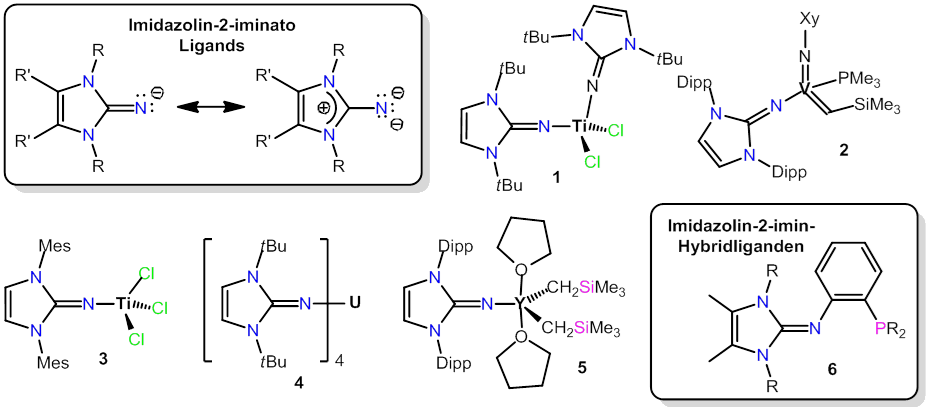
In addition to these anionic ligands, we employ (poly)imidazolin-2-imines for applications in coordination, medicinal and bioinorganic chemistry [1]. Future work will comprise, inter alia, hybride ligands such as 6, which will be used for the development of catalysts for hydrogen transfer catalysis, electrocatalysis (H2 oxidation) and CO2 hydrogenation.
[1] Reviews: "Transition metal complexes supported by highly basic imidazolin-2-iminato and imidazolin-2-imine N-donor ligands", Coord. Chem. Rev. 2014, 260, 116-138; "Rare Earth Metal Complexes Supported by Ancillary Imidazolin-2-iminato Ligands", Z. Anorg. Allg. Chem. 2010, 636, 2156-2171.
[2] "Bis(1,3-di-tert-butylimidazolin-2-iminato) Titanium Complexes as Effective Catalysts for the Monodisperse Polymerization of Propylene", J. Am. Chem. Soc. 2012, 134, 17234-17244.
[3] "Synthesis and Structural Analysis of (Imido)vanadium(V) Dichloride Complexes Containing Imidazolin-2-iminato- and Imidazolidin-2-iminato Ligands, and their Use as Catalyst Precursors for Ethylene (Co)polymerization", Inorg. Chem. 2014, 53, 607-623.
[4] "Mono(imidazolin-2-iminato) Titanium Complexes for Ethylene Polymerization at Low Amounts of Methylaluminoxane", J. Am. Chem. Soc. 2013, 135, 12592-12595.
[5] "Uranium(IV) Imidazolin-2-iminato Complexes: A New Class of Organoactinides", Inorg. Chem. 2014, 53, 694-696.
[6] "Bis(imidazolin-2-iminato) Rare Earth Metal Complexes: Synthesis, Structural Characterization, and Catalytic Application", Inorg. Chem. 2012, 51, 6753-6761; "Syntheses and Structures of Mononuclear Lutetium Imido Complexes with Very Short Lu-N Bonds", Chem. Commun. 2007, 5007-5009.
Catalytic alkyne metathesis involving the reversible cleavage and formation of carbon-carbon triple bonds still receives relatively little attention in comparison with the related Nobel prize-winning olefin metathesis technology [1]. Based on a design concept that draws on the strucutre of highly active olefin metathesis catalysts of the Schrock-type, we obtained molybdenum and tungsten alkylidyne complexes of type 1 with ancillary imidazolin-2-iminato ligands, and these catalysts were able to efficiently promote alkyne metathesis even at room temperature and low catalyst loadings [2]. Other classes of highly active catalysts comprise alkylidyne complexes with phosphoraniminato (2) and silanolate ligands (3) [3,4]. Surprsingly, we have observed that the latter species are able to catalyse the metathesis of conjugated diynes,[5] and we are convinced that these reactions will have a great potential for application in organic synthesis and materials sience. The molybdenum complex 4 represents the most active alkyne metathesis catalyst to date, and it even allowed - for the first time - the efficient metathesis of terminal alkynes [6]. In addition to further development and application of these catalysts, future work will also strongly focus on mechanistic studies of alkyne and diyne metathesis.
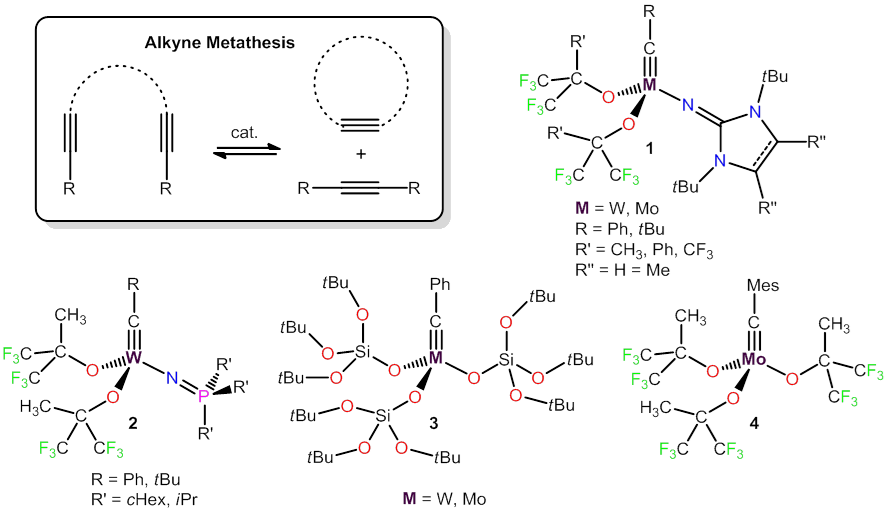
[1] Review: "Recent advances in the development of alkyne metathesis catalysts", Beilstein J. Org. Chem. 2011, 7, 82-93.
[2] "Efficient Room-Temperature Alkyne Metathesis with Well-Defined Imidazolin-2-iminato Tungsten Alkylidyne Complexes", Angew. Chem. 2007, 119, 9047-9051; Angew. Chem. Int. Ed. 2007, 46, 8890-8894; "Preparation of Imidazolin-2-iminato Molybdenum and Tungsten Benzylidyne Complexes - A New Pathway to Highly Active Alkyne Metathesis Catalysts", Chem. Eur. J. 2010, 16, 8868-8877.
[3] "Efficient Catalytic Alkyne Metathesis with a Tri(tert-butoxy)silanolate-Supported Tungsten Benzylidyne Complex", ChemCatChem 2011, 3, 115-118.
[4] "Tungsten and Molybdenum 2,4,6-Trimethylbenzylidyne Complexes as Robust Pre-Catalysts for Alkyne Metathesis", Adv. Synth. Catal. 2014, 356, 1255-1265.
[5] "Catalytic Metathesis of Conjugated Diynes", Angew. Chem. 2012, 124, 6861-6865; Angew. Chem. Int. Ed. 2012, 51, 6757-6761; "Synthesis of Unsymmetrical 1,3-Diynes via Alkyne Cross-Metathesis", Chem. Commun. 2013, 49, 7189-7191.
[6] "Efficient Metathesis of Terminal Alkynes", Angew. Chem. 2012, 124, 13195-13199; Angew. Chem. Int. Ed. 2012, 51, 13019-13022.
The reaction between an electron-pair acceptor (Lewis acid) and an electron-pair donor (Lewis base) is one of the fundamental chemical processes and usually results in the formation of an acid-base adduct. Using sterically demanding acids and bases may suppress adduct formation and generate a so-called "frustrated Lewis pair" (FLP), whose unrestrained Lewis acidity and Lewis basicity remain available for the mutual activation of suitable molecules. Our contribution to this field comprise the use N-heterocyclic carbene-borane Lewis pairs of the type 1/2, which accomplish the heterolytic cleavage of dihydrogen, stoichiometric bond activation (C-H, N-H, Si-H), dehydrogenation as well as activation and fixation of small molecules [1,2]. Thereby, the reactivity of these FLP systems can be modulated by variation of the carbene and borane components [3]. In addition, we developed several intramolecular FLPs such as the pyrazolylborane 3 for stoichiometric and catalytic applications [4].
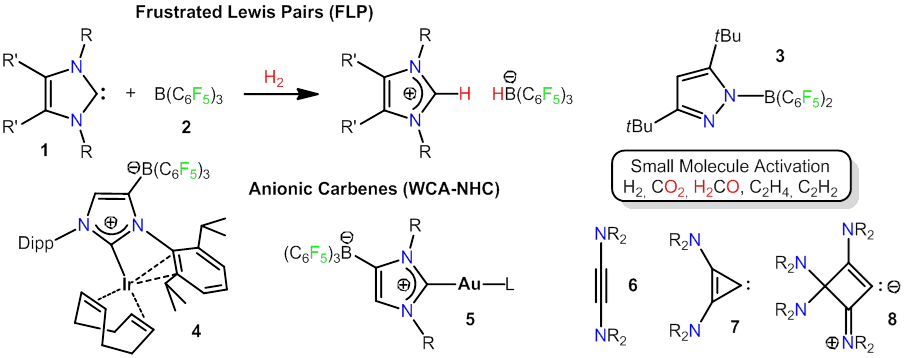
As a spin-off from our FLP chemistry, we introduced anionic N-heterocyclic carbenes with a weakly coordnating borate moiety (WCA-NHC), which were used for the development of homogeneous catalysts. Thus, neutral iridium(I) and gold(I) complexes 4 and 5 were prepared as uncharged analogues of well established cationic catalysts and used for catalytic hydrogenation and skeletal rearrangements [5,6]. Other fields of interest cover diaminoacetylenes (6) and formally derived carbenoid three- and four-membered ring species such as 7 und 8 [7,8].
[1] Review: "N-Heterocyclic Carbenes in FLP Chemistry", Top. Curr. Chem. 2013, 334, 121-155.
[2] "Heterolytic H2 Activation by a Frustrated Carbene-Borane Lewis Pair", Angew. Chem. 2008, 120, 7538-7542; Angew. Chem. Int. Ed. 2008, 47, 7428-7432.
[3] "Dihydrogen activation by frustrated carbene-borane Lewis pairs: An experimental and theoretical study of carbene variation", Inorg. Chem. 2011, 50, 7344-7359; "Reactivity of a Frustrated Lewis Pair and Small-Molecule Activation by an Isolable Arduengo Carbene-B{3,5-(CF3)2C6H3}3 Complex", Chem. Eur. J. 2012, 52, 16938-16946 .
[4] "Intramolecular heterolytic dihydrogen cleavage by a bifunctional frustrated pyrazolylborane Lewis pair", Chem. Commun. 2010, 46, 8561-8563.
[5] "Anionic N-Heterocyclic Carbenes That Contain a Weakly Coordinating Borate Moiety", Angew. Chem. 2012, 124, 3294-3298; Angew. Chem. Int. Ed. 2012, 51, 3240-3244.
[6] "Iridium(I) Complexes with Anionic N-Heterocyclic Carbene Ligands as Catalysts for the Hydrogenation of Alkenes in Non-Polar Media", J. Am. Chem. Soc. 2013, 135, 12448-12459.
[7] "A Novel Synthetic Approach to Diaminoacetylenes: Structural Characterization and Reactivity of Aromatic and Aliphatic Ynediamines", Chem. Eur. J. 2010, 16, 11804-11808.
[8] "A Stable Chiral Diaminocyclopropenylidene", Chem. Commun. 2007, 3661-3663.
In comparsion with cyclopentadienyl and arene ligands, their seven-membered cycloheptatrienyl congener can be regarded as a neglected carbocyclic ligand system in organometallic chemistry. With our contribution, we aimed at changing this situation and have drawn our attention to cycloheptatrienyl-cyclopentadienyl (Cht-Cp) sandwich complexes [1]. The reactivity of strained ansa-Cht-Cp complexes such as 1 was studied and exploited for the synthesis of metallopolymers by ring-opening polymerisation [2]. Phosphane ligands of type 2 were particularly interesting for catalytic applications, since these 16-electron complexes are able to develop secondary interactions involving the metal atoms [3]. The readily accessible titanium complexes ("troticenyl phosphanes")[4] could be applied as reducing phosphanes in palladium-catalysed Suzuki-Miyaura cross-couplings, with the troticene complex fragment acting as the reducing agent for rapid formation of catalytically active Pd(0) species [5]. Further application of these ligands, which have already caught the attention of several chemical companies, in homogeneous catalysis is currently thoroughly investigated.
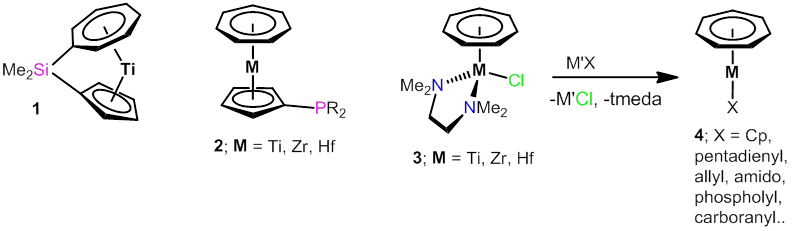
A substantial part of our organometallic chemistry with seven-membered rings is dedicated to the development of readily accessible starting materials for the preparation of novel Cht complexes. Thus, half-sandwich complexes such as 3 are suitable precursors for the synthesis of numerous heteroleptic complexes of type 4, which can be employed for subsequent stoichiometric and catalytic transformations [6,7]. These studies were mainly carried out with the zirconium derivative and will henceforth be transferred to the corresponding titanium and hafnium compounds.
[1] Reviews: "The Organometallic Chemistry of Cycloheptatrienyl Zirconium Complexes", Chem. Soc. Rev. 2013, 42, 128-142; "Synthesis and Reactivity of Functionalized Cycloheptatrienyl-Cyclopentadienyl Sandwich Complexes", Chem. Commun. 2008, 3089-3100.
[2] "Ansa-Cycloheptatrienyl-Cyclopentadienyl Complexes", Angew. Chem. 2004, 116, 5646-5650; Angew. Chem. Int. Ed. 2004, 43, 5530-5534; "Regioselective Si-C bond Activation in Silicon-Bridged Ansa-Cycloheptatrienyl-Cyclopentadienyl Complexes", Chem. Commun. 2005, 1729-1731; "Synthesis and Reactivity of Boron-, Silicon- and Tin-Bridged ansa-Cyclopentadienyl-Cycloheptatrienyl Titanium Complexes (Troticenophanes)", Chem. Eur. J. 2010, 16, 11732-11743.
[3] "Secondary Interactions in Phosphane-Functionalized Group 4 Cycloheptatrienyl-Cyclopentadienyl Sandwich Complexes", Chem. Eur. J. 2009, 15, 2176-2184.
[4] "Selective Lithiation and Phosphane-Functionalization of [(η7-C7H7)Ti(η5-C5H5)] (Troticene) and Its Use for the Preparation of Early-Late Heterobimetallic Complexes", J. Am. Chem. Soc. 2009, 131, 17014-17023.
[5] "Phosphane-Functionalized Cycloheptatrienyl-Cyclopentadienyl Titanium Sandwich Complexes: Phosphorus Ligands with an Integrated Reducing Agent for Pd(0) Catalyst Generation", Angew. Chem. 2013, 125, 8800-8804; Angew. Chem. Int. Ed. 2013, 52, 8638-8642
[6] "Cycloheptatrienyl Zirconium Sandwich Complexes with Lewis Basic Phospholyl Ligands (Phosphatrozircenes) - Synthesis, Structure, Bonding and Coordination Chemistry", Chem. Eur. J. 2011, 17, 6118-6128.
[7] "A slipped multi-decker zirconium complex with an η7:η2 bridging cycloheptatrienyl ligand", Chem. Commun. 2012, 48, 6598-6600.